How to produce
infarction in the mouse in vivo
Yiru Guo, M.D.
Xian-Liang Tang, M.D.,
Weike Bao, M.D.,
Roberto Bolli, M.D
From the Experimental Research Laboratory
Division of Cardiology
University of Louisville and Jewish Hospital
Heart and Lung Institute
Louisville, KY 40292
Tel (502) 852 1837
Fax (502) 852 6474
E-mail: rbolli@louisville.edu
INTRODUCTION
The mouse has emerged as a popular
experimental species because of the increasing availability of
transgenic and gene targeted strains that are now becoming
important in the study of cardiovascular disease. Therefore,
characterizing an applicable model of myocardial infarction (MI)
in the mouse is important to test different hypotheses by
utilizing transgenic and gene targeted mice, wherein the
manipulation of specific genes may allow further mechanistic
insights. For example, the finding that ischemic preconditioning
cardioprotection against MI is abrogated in mice homozygous for a
null iNOS allele (iNOS-/-) has furnished
new insights into the pathophysiology of ischemic injury.
The mouse is likely to continue to be the
species commonly used for transgenesis and gene targeting.
Although transgenesis is theoretically possible in larger mammals
(e.g., rabbits or pigs), developing such models would be
extremely costly and time-consuming, making these models
impractical. Furthermore, gene targeting has not been reported
thus far in species other than the mouse. The development of a
physiologically-relevant mouse model of myocardial infarction is
a necessary step towards the utilization of
genetically-engineered animals for interrogating the molecular
basis of myocardial ischemia/reperfusion injury and
preconditioning. The unique technical challenges associated with
inducing MI in mice raise the concern that the results obtained
in this model may not be as reliable and physiological as those
obtained in larger species, where the margin for error is wider
and physiologic parameters can be measured more easily. To
thoroughly address these concerns, we have developed a murine
model of myocardial infarction in which fundamental physiologic
variables (body temperature, heart rate, arterial blood pressure,
arterial blood gases and pH) are carefully measured and
controlled.1,2 We have shown that measurements of infarct size in
this model are both reliable and reproducible. Furthermore, we
have found that both the early and the late phases of ischemic
preconditioning are robustly expressed in the murine model.1,2 Utilizing this model, we have shown that
the inducible isoform of NOS is essential for the late phase of
preconditioning induced by ischemia or by pharmacologic
stimulation with adenosine A1 agonists or opioid
agonists.3We have also demonstrated that overexpression of
PKCe confers protection against infarction and that targeted
deletion of Lck blocks late preconditioning. Thus, this model
appears to be a useful tool to interrogate the molecular basis of
ischemia/reperfusion injury with a degree of specificity that is
not possible with pharmacologic approaches in other species.
Accordingly, the purpose of this chapter is to describe the
methodological details that are employed for measuring infarct
size in our murine model.
What mouse
strain do we use?
In general, we prefer to use adult male
mice. We think the best strains are ICR (outbred), C57BL/6J
(inbred), and B6129F2/J (hybrid). These strains have good
reproductive performance and also preconditionable.1,2,4
The ICR strain is a hardy outbred
stock developed at the Institute for Cancer Research (Fox Chase
Cancer Center) by T.S. Hauschka beginning in 1948. Good
reproductive performance and fast growth rate characterize this
strain. It has been used extensively in toxicology and
pharmacology studies. It is often used for product safety testing
and as an embryo donor and/or recipient mother in transgenic
mouse labs. ICR mice can be obtained from Harlan Sprague Dawley
(Houston, TX) or Taconic (Germantown, NY).
The C57BL/6J strain, one of the
C57BL substrains, is now probably the most widely used of all
inbred strains. It is popular in research applications of
oncology and toxicology and is used as the female parents to
produce the B6129F1, B6C3F1, and B6D2F1 hybrids mice. The mice
can be purchased from Jackson Laboratory (Bar
Harbor, ME) or Taconic (Germantown,
NY).
The B6129F2/J has two stocks (B6129SF2/J
and B6129PF2). In general, an F2 hybrid is defined as
the second filial generation and is produced by mating two F1
hybrid mice. Matings to produce F2 mice provide the opportunity
for genetic recombination between all differing loci to occur.
Therefore, F2 hybrids are not genetically identical. For example,
if strain X (which carries an 'a' allele at all loci) is mated
with strain Y (which carries a 'b' allele at all loci), F2
progeny could be a/a, a/b, or b/b at each individual locus. An F1
hybrid is defined as the first filial generation, or the
offspring of an outcross between two different strains. F1
hybrids are heterozygous at all loci at which their parents have
different alleles. Similar to inbred strains, F1 hybrids are
genetically and phenotypically uniform. Continuing with the
example above where strain X and strain Y were mated, the F1
progeny would be a/b at every loci. Additionally, F1 hybrids
exhibit 'hybrid vigor': increased disease resistance, better
survival under stress, greater natural longevity, larger litters,
and so on. Two other uses of F1 hybrids include:
1) accepting transplants of tissues from
mice of either parental strain.
2) used as backgrounds for some deleterious
mutations.
An F1 hybrid would be used in situations
where genetic homogeneity is important; F2 hybrids might be used
in situations where genetic homogeneity is less important, or in
situations where they best approximate the mixed genetic
background of experimental mice.
Recently, we have found that the
genetic strain is an important determinant of the susceptibility
of mice to myocardial infarction and ischemic preconditioning.4 Specifically, we have found that FVB/N
mice have much smaller infarcts after a 30-min coronary occlusion
than do the ICR, C57BL/6J, and B6/129SF2/J mice, and that 129SvEv
mice do not develop late preconditioning.4Thus, when using gene targeted or transgenic mice,
controls should consist of mice with a genetic background as
close as possible to that of the gene targeted or transgenic mice
(littermates are the best controls, if they are available). The
important concept is that data obtained in one strain of mice
cannot be extrapolated to another strain of mice.
How do we house the mice?
The mouse should be housed in microisolator
cages under specific pathogen-free conditions in a room with a
temperature of 24C, 55%-65% relative humidity, and a 12-h
light-dark cycle. For maintaining pathogen-free transgenic or
gene target mice, we generally keep mice in sterile microisolator
cages under pathogen-free conditions, and food and water are
autoclaved and all handling is done under a laminar flow hood
following standard procedures. We allow the mice to acclimate to
our facility for >2 wks before experimentation (because
shipping may cause some stress). However, mice exhibiting
symptoms of illness such as weight loss, decreased food intake,
hair loss, loss of vigor, or wounds from fighting are excluded
from the study before the beginning of the experiment.
How do we perform the open-chest
procedure?
Mice are premedicated with atropine sulfate
(0.04 mg/kg i.m.) and anesthetized 5 min later with sodium
pentobarbital (50 mg/kg i.p.). Additional doses of pentobarbital
are given during the protocol as needed to maintain anesthesia.
The animals are placed in a supine position with the paws taped
to the operating table. Surface leads are placed subcutaneously
to obtain the ECG, which is recorded throughout the experiments
on a thermal array chart recorder. Before starting surgery, mice
are given gentamicin (0.7 mg/kg i.m.).
A midline cervical skin incision is
performed and the muscles overlying the trachea are reflected to
allow visualization of the endotracheal tube (a PE-60~90 tubing)
as it is placed in the trachea. To facilitate intubation, a
rubber band is placed behind the upper incisors and fastened to
the operating table so that the neck is slightly extended. To
place the endotracheal tube, the tongue is slightly retracted,
and the beveled end of the tube (which is marked with a black
marker) is inserted carefully through the larynx and into the
trachea so as not to puncture the trachea or other structures in
the pharyngeal region. The tube is advanced 8-10 mm from the
larynx and taped in place to prevent dislodgment. The animals are
ventilated with room air supplemented with oxygen (2 L/min) at a
rate of 105/min and with a tidal volume of 2.1-2.5 ml using a
rodent ventilator (Harvard Apparatus,
Inc., South Natick, MA). The endotracheal tube is inserted
loosely into the tube connected to the ventilator so as to avoid
lung overexpansion. A catheter is inserted into the external
jugular vein for fluid infusion. In selected studies, a
catheter is inserted into the carotid artery for measurement of
blood pressure (DTXTM pressure transducer,
Viggo-Spectramed, Oxnard, CA) and analysis of blood gases.
To replace blood losses, blood from a donor mouse is given i.v.
at a dose of 40 ml/kg (~1 ml) divided into three equal boluses
(first bolus, after connecting the endotracheal tube to the
ventilator; second bolus, after opening the chest; third bolus,
after closing the chest). Body temperature is carefully
monitored with a rectal probe connected to a digital thermometer
(Cole-Parmer Instrument Company,
Vernon Hill, IL) and is maintained as close as possible to 37.0 C
throughout the experiment using a heating pad and heat lamps.
How do we perform the coronary
artery occlusion procedure?
With the aid of a dissecting microscope (Fisher Scientific,
Pittsburgh, PA) and a microcoagulator (ASSI Polar-Mate Isolator,
San Diego, CA), the chest is opened through a midline
sternotomy. An 8-0 nylon suture (Ethicon, Inc. Johnson
& Johnson Co. Somerville, NJ) is passed with a tapered needle
under the left anterior descending coronary artery 2-3 mm from
the tip of the left auricle, and a nontraumatic balloon occluder
is applied on the artery. Coronary occlusion is induced by
inflating the balloon occluder. Successful performance of
coronary occlusion and reperfusion is verified by visual
inspection (i.e., by noting the development of a pale color in
the distal myocardium upon inflation of the balloon and the
return of a bright red color due to hyperemia after deflation)
and by observing ST-segment elevation and widening of the QRS on
the ECG during ischemia and their resolution after
reperfusion. After the coronary occlusion/reperfusion
protocol, the chest is closed in layers, and a small catheter is
left in the thorax for 10-20 min to evacuate air and
fluids. The mice are removed from the ventilator, kept warm
with heat lamps, given fluids
(1.0-1.5 ml of 5% dextrose in water i.p.), and allowed 100%
oxygen via nasal cone.
How do we perform postmortem
analysis?
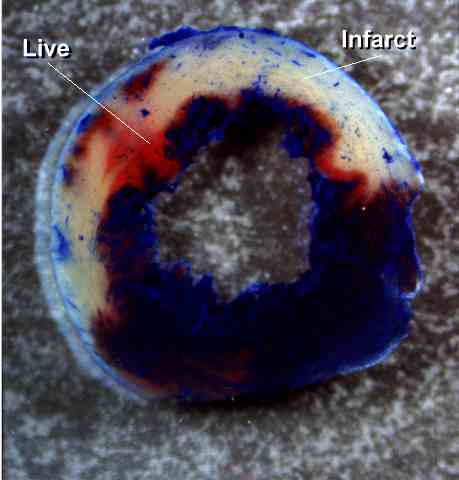 Figure
1. Representative example of a heart from a
control group (ICR subjected to a 30-min coronary occlusion and
24 h of reperfusion). The infarcted region was delineated
by perfusing the aortic root with TTC; the region at risk was
delineated by perfusing the aortic root with Phthalo blue after
tying the previously occluded artery (see text for
details). As a result of this procedure, the nonischemic
portion of the left ventricle was stained dark blue, the viable
tissue within the region at risk was stained bright red, whereas
the infarcted tissue was light yellow. Note the large,
confluent areas of infarction spanning most of the thickness of
the LV wall, with thin rims of viable subendocardial
tissue. This pattern was characteristic of all
nonpreconditioned groups (including AMI and sham groups).
The scale at the bottom is in mm.
Figure
1. Representative example of a heart from a
control group (ICR subjected to a 30-min coronary occlusion and
24 h of reperfusion). The infarcted region was delineated
by perfusing the aortic root with TTC; the region at risk was
delineated by perfusing the aortic root with Phthalo blue after
tying the previously occluded artery (see text for
details). As a result of this procedure, the nonischemic
portion of the left ventricle was stained dark blue, the viable
tissue within the region at risk was stained bright red, whereas
the infarcted tissue was light yellow. Note the large,
confluent areas of infarction spanning most of the thickness of
the LV wall, with thin rims of viable subendocardial
tissue. This pattern was characteristic of all
nonpreconditioned groups (including AMI and sham groups).
The scale at the bottom is in mm.
At the conclusion of the study,
the mice are given heparin (1 U/g i.p.), after which they are
anesthetized with sodium pentobarbital (35 mg/kg i.p.) and
euthanized with an i.v. bolus of potassium chloride
(KCl). The heart is excised and perfused with
Krebs-Henseleit solution through an aortic cannula (22- or
23-gauge needle) using a Langendorff apparatus. To
delineate infarcted from viable myocardium, the heart is then
perfused with a 1% solution of triphenyltetrazolium chloride
(TTC) in phosphate buffer (pH 7.4, 37 C) at a pressure of 60 mmHg
(approximately 3 ml over 3 min). To delineate the
occluded/reperfused coronary vascular bed (the region at risk),
the coronary artery is then tied at the site of the previous
occlusion and the aortic root is perfused with a 5-10% solution
of Phthalo blue dye (Heucotech Ltd., Fairless Hill, Pa) in normal saline (2 ml over
3 min). As a result of this procedure, the portion of the
left ventricle (LV) supplied by the previously occluded coronary
artery (region at risk) is identified by the absence of blue
dye, whereas the rest of the LV is stained dark blue. The
heart is frozen, after which all atrial and right ventricular
tissues are excised. The LV is cut into 5-7 transverse
slices, which are fixed in 10% neutral buffered formaldehyde and,
24 h later, weighed and photographed (Nikon AF N6006). The
transparencies are projected onto a paper screen at a 30-fold
magnification, and the borders of the infarcted,
ischemic-reperfused, and nonischemic regions are traced.
The corresponding areas are measured by computerized
videoplanimetry (Adobe Photoshop, version 4.0, NIH Image, or
Image tool), and from these measurements infarct size is
calculated as a percentage of the region at risk.1,2
What kind of anesthetic do we use?
Initially, we induced anesthesia with
xylazine (7.5 mg/kg i.m.) and ketamine (55 mg/kg i.m.); however,
we found that the heart rate was quite low (280-330 bpm), not
within the physiological range. We therefore chose
pentobarbital anesthesia. After selecting the anesthetic, a
series of pilot studies was performed in 47 mice.2
First, we sought to establish physiological parameters to be used
as a reference for subsequent experiments. In eight mice,
ECG leads were placed subcutaneously and the animals were allowed
to recover. Heart rate was monitored in the conscious state
on the following days, and was found to average 668±31 bpm
(range, 490 to 760 bpm). In 16 pentobarbital-anesthetized
mice, mean arterial blood pressure prior to thoracotomy was found
to average 97.2±4.4 mmHg. In 39 pentobarbital-anesthetized
mice, rectal temperature prior to thoracotomy was found to
average 37.0±0.2°C. Therefore, under pentobarbital anesthesia,
heart rate and arterial pressure are within physiological
limits. We have found that mice recover fairly quickly
after closing the thoracotomy, and believe that pentobarbital
provides a satisfactory means to induce anesthesia.
What ventilatory parameters do we use?
Because of the obvious importance of
adequate oxygenation and acid-base balance, we recommend that
investigators identify the optimal ventilatory parameters in
their particular model. In our studies, since the
endotracheal tube used is without a cuff, we adjust the tidal
volume by observing the inflation of the lungs after the chest is
opened. We found that the average tidal volume of 2.2 +/-
0.1 ml results in adequate inflation of the lungs without
over-expansion. (Over-expansion is dangerous because it can cause
rupture of the lungs) Using this tidal volume, we tested
different ventilatory rates in 23 open-chest mice and analyzed
arterial blood gases in each animal (Table 1).2
We found that even small changes in ventilatory rate resulted in
significant changes in arterial blood gases (Table 1), emphasizing
the importance of the ventilatory parameters. A ventilatory
rate of 105/min was found to produce optimal values of pO2,
pCO2, and pH, as detailed in Table 1. This
rate is within the range observed in spontaneously breathing
mice.5,6 The ventilatory rate and the tidal volume that
produce the optimal blood gas values will vary from one
laboratory to another. What is important is that each
laboratory determines these values before starting large-scale
studies of myocardial infarction in mice.
How can arterial blood pressure be kept
in the normal range during the occlusion/reperfusion procedure?
We measured arterial blood
pressure in mice subjected to open-chest surgery.2 The results are summarized in Fig 2.
In five mice, one dose of blood (13.3 ml/kg; ~0.4 ml) was given
immediately after the thoracotomy in an effort to prevent
hypotension. Despite this, mean arterial blood pressure
fell to 63.4±3.7 mmHg after opening the chest (probably due to
the loss of negative intrathoracic pressure and to the positive
end-expiratory pressure) (Fig. 2). Furthermore, after closing the chest,
another drop in blood pressure was noted, to a nadir of 63.0±9.7
mmHg (Fig. 2). Because these hypotensive episodes could induce
ischemic preconditioning, we decided to administer three doses of
blood (instead of one): the first was given before opening the
chest, the second immediately after opening the chest, and the
third after closing the chest. Each dose consisted of 13.3
ml/kg (~0.4 ml). Using this protocol, mean arterial
pressure remained at or above 80 mmHg throughout a 1-h period of
open-chest state (Fig. 2). Next, we tested whether this protocol of
fluid replacement was sufficient to prevent severe hypotension in
mice undergoing a sequence of six coronary occlusion/reperfusion
cycles, in which myocardial ischemia would be expected to cause a
further fall in arterial pressure. Although each coronary
occlusion caused a drop in arterial pressure, the three doses of
blood resulted in mean arterial pressure being maintained at or
above 80 mmHg throughout the six occlusion/reperfusion cycles (Fig. 2). Thus, using the fluid supplementation
protocol detailed above and careful precautions to minimize blood
loss, arterial blood pressure could be kept at adequate levels
throughout the six occlusion/reperfusion cycles.
How can body temperature and
heart rate be maintained within the physiologic range?
Of all the determinants of infarct
size, temperature is probably the most important.
Accordingly, temperature should be strictly controlled throughout
the experiment. We control temperature by adjusting the
heating pad and the heat lamps during the surgical
procedures. We allow a minimal drift in temperature, from
36.7° to 37.3°C. As a result of these compulsive
measures, rectal temperature remains within the physiological
range in our studies (Table 2). With regard to heart rate, as mentioned
earlier, pentobarbital anesthesia results in heart rates those
are within the physiologic range for conscious mice (this range
is from 500 to 760 beats/min) (Table 2).
How to select the duration of
reperfusion?
In view of the added complexity
inherent in following mice for 24 h after reperfusion, we
investigated whether extending the reflow period beyond 4 h was
important for assessing the final extent of infarct size.
Birnbaum et al.7 have reported that at least 3
h of reperfusion are necessary to assess the final extent of
infarction in rabbits. Accordingly, we allowed a minimum of
4 h of reperfusion. When infarct size was compared after 4
h and 24 h of reperfusion, the results were similar, both in
nonpreconditioned hearts (group II vs. group I; group VII vs.
group V) and in preconditioned hearts (group VIII vs. group VI) (Fig. 3 and Table 3), supporting
the conclusion that a 4-h reperfusion interval is sufficient to
evaluate ischemic preconditioning in the mouse.2
This information should be useful in designing future studies,
particularly studies of early preconditioning, which could be
done acutely without the need for survival surgery.2
Although the results obtained with 4 h and 24 h of reperfusion
are similar, we think it is preferable, whenever possible, to
allow 24 h of reperfusion, particularly when using mice with
genetic mutations of proteins that may be important in modulating
the inflammatory response after reperfusion. However, when
mice are particularly frail, a 4-h reperfusion interval is
preferable because it reduces mortality.
DISCUSSION
We have developed a reliable and
physiologically-relevant model of ischemic preconditioning that
can be used in genetically-engineered animals. Our results
can be summarized as follows (i) despite the small size of the
mouse, it is possible to study MI in vivo in this model under
conditions in which basic physiologic variables are kept within
normal limits (body temperature, arterial oxygenation, acid-base
balance, heart rate, and arterial blood pressure); and (ii) both
the quality of the postmortem staining for region at risk and
infarction and the reproducibility of the measurements of infarct
size are excellent and compare favorably with those in larger
species.
This murine model of MI should be useful
for investigating the impact of genetic manipulations upon
physiological end-points in vivo. By applying this
model to mice with overexpression or targeted disruption of
individual genes implicated in the cellular pathways underlying
ischemic heart diseases, it should be possible to conclusively
establish the role of a specific gene product in the genesis of
ischemic heart diseases in the intact animal.
A major concern in the design of our model
was to ensure that the results would be physiologically
relevant. The minuscule size of the murine heart
necessitates miniaturization of the procedures used in larger
species and therefore poses a unique challenge in terms of
maintaining general experimental conditions within normal values
and avoiding artifacts. Thus, when starting a mouse model,
a considerable amount of preliminary work should be performed
prior to the actual studies.
Because temperature is a major determinant
of infarct size,8-11 this variable is tightly controlled throughout the
experiment by using heating pads and heat lamps while
continuously monitoring rectal temperature. Our results
demonstrate that by using these procedures, temperature can be
kept within a narrow range (36.4-37.6°C) (Table 2), which
represents the normal range for the mouse,5,12 as confirmed by our pilot studies in which rectal
temperature averaged 37.0°±0.4°C. Hypoxemia, acidosis,
and alkalosis may also have a major influence upon animal
survival, infarct size, and/or ischemic preconditioning.
Accordingly, we measured arterial pH, pO2, pCO2,
and bicarbonate levels in mice subjected to open-chest surgery (Table 1). These
measurements demonstrated that, with a ventilatory rate of
105/min and an average tidal volume of 2.2 ml, all parameters
were within the physiologic range for the mouse13; in particular, arterial pH was kept at ~7.40 and
adequate oxygenation was maintained throughout the open-chest
state (Table 1). Careful
control of blood gases is important in the mouse, since small
variations in ventilatory rate result in marked variations in
arterial blood gases (Table 1).
Heart rate and arterial pressure are
important indices of normal cardiovascular homeostasis and are
also important determinants of the severity of myocardial
ischemia. As elaborated earlier, we avoided anesthesia with
ketamine/xylazine because these agents resulted in unacceptably
low heart rates (280 to 330 bpm), clearly outside of the
physiological range, which in our pilot studies in conscious mice
was found to be 490 to 760 bpm (average, 688±31 bpm). With
pentobarbital anesthesia, the heart rates recorded in the present
experiments (Table 2) were
reasonably close to those measured in conscious mice in our pilot
studies and in previous studies.6,12,14,15
The blood volume of a 25-g mouse has been estimated to range
between 1.5 and 2.3 ml.5 To avoid hypotension, surgery was
performed with a microcoagulator, and every effort was made to
minimize blood losses. Pilot studies, however, showed that
opening the chest caused a significant drop in arterial blood
pressure, so that the mice became severely hypotensive despite
the administration of 13.3 ml/kg (~0.4 ml) of blood (Fig. 2).
Besides causing mortality, severe hypotension could lead to
myocardial hypoperfusion and, possibly, induce preconditioning as
a result of myocardial ischemia and/or reflex adrenergic
activation. We therefore modified our protocol by
administering three doses of blood (total of 40 ml/kg or ~1.2
ml), as detailed earlier, which resulted in values of mean
arterial blood pressure >80 mmHg throughout the sequence of
six coronary occlusion/reperfusion cycles (Fig. 2). These
values of arterial pressure are within the range reported by
others in normal mice.5,14-22
Using this model, the average infarct size
in nonpreconditioned mice (groups I, II, III, V and VII combined,
see Fig. 3) was found to
be 52% of the region at risk, which, surprisingly, is similar to
the average infarct size measured after the same duration of
coronary occlusion (30 min) in conscious rabbits (56.9±5.9% 23and 56.8±5.3% 24of the
region at risk) and in open-chest rabbits (52.0±5.2% 25,
53.6±5.7% 26, 48.1±3.9% 27, and
49.1±4.3% 28 of the region at risk). The ranges
of individual infarct sizes (Fig. 3 and Table 3) and the
slopes and x-intercepts of the infarct size-risk region
relationships (Fig. 4) were also
similar to those previously observed in conscious rabbits after a
30-min occlusion.23,24
CONCLUDING COMMENTS
With the development of
genetically-engineered mice, there is increasing interest in
using transgenic or knockout mice as a tool to interrogate the
cellular mechanisms of cardiovascular disease. By
overexpression or targeted disruption of specific genes, these
murine models provide a unique approach to understanding the role
of specific gene products in abnormal cardiovascular
function. In the case of ischemic preconditioning, however,
the exploitation of genetic manipulations has been hindered by
the lack of in vivo physiologic correlates. We have
developed a mouse model of myocardial infarction in which several
fundamental physiological variables are carefully controlled and
kept within normal limits. Mortality is relatively low
(<20%). Measurements of infarct size are accurate and
reproducible. Our results also demonstrate that a robust
infarct-sparing effect occurs during the early and the late
phases of preconditioning in the mouse and that the quantitative
aspects of this effect are consistent with previous experience in
other species. This model has been useful to elucidate the
molecular basis of ischemic preconditioning by making it possible
to apply molecular biology techniques to intact animal
preparations in order to dissect the precise roles of individual
proteins.
REFERENCES
1. Guo Y, Jones WK, Xuan YT, Tang XL, Bao
W, Wu WJ, Han H, Laubach VE, Ping P, Yang Z, Qiu Y, Bolli R. The
late phase of ischemic preconditioning is abrogated by targeted
disruption of the inducible NO synthase gene [see comments]. Proc
Natl Acad Sci U S A. 1999;96:11507-11512.
2. Guo Y, Wu WJ, Qiu Y, Tang XL, Yang Z,
Bolli R. Demonstration of an early and a late phase of ischemic
preconditioning in mice. Am J Physiol.
1998;275:H1375-1387.
3.Guo Y, Bao W, Tang XL, Wu WJ, Takano H,
Bolli R. Activation of adenosine A1 and d1opioid
receptors induces late preconditioning in mice. J Mol Cell
Cardiol. 2000;32:A51.
4. Bao W, Guo Y, Tang XL, Wu WJ, Bolli R.
Variability in susceptibility to ischemia among different strains
of mice. J Mol Cell Cardiol. 2000;32:A21.
5. Kaplan H, Brewer N, Blair W. The
cardiovascular system. Normative biology, immunology, and
husbandry. New York: Academic; 1983.
6. Wolfensohn S, Lloyd M. Biological
data. Oxford, UK: Oxford University Press; 1996.
7.Birnbaum Y, Hale SL, Kloner RA.
Differences in reperfusion length following 30 minutes of
ischemia in the rabbit influence infarct size, as measured by
triphenyltetrazolium chloride staining. J Mol Cell Cardiol.
1997;29:657-666.
8.Chien GL, Wolff RA, Davis RF, van Winkle
DM. "Normothermic range" temperature affects myocardial
infarct size. Cardiovasc Res. 1994;28:1014-1017.
9. Duncker DJ, Klassen CL, Ishibashi Y,
Herrlinger SH, Pavek TJ, Bache RJ. Effect of temperature on
myocardial infarction in swine. Am J Physiol.
1996;270:H1189-1199.
10. Hale SL, Kloner RA. Myocardial
temperature in acute myocardial infarction: protection with mild
regional hypothermia. Am J Physiol. 1997;273:H220-227.
11. Schwartz LM, Verbinski SG, Vander Heide
RS, Reimer KA. Epicardial temperature is a major predictor of
myocardial infarct size in dogs. J Mol Cell Cardiol.
1997;29:1577-1583.
12. Siegmund OT. Some physiologic
values. Rahway, NJ: Merk; 1979.
13. Erhardt W, Hebestedt A, Aschenbrenner
G, Pichotka B, Blumel G. A comparative study with various
anesthetics in mice (pentobarbitone, ketamine-xylazine,
carfentanyl-etomidate). Res Exp Med. 1984;184:159-169.
14. Schlager G. Selection for blood
pressure levels in mice. Genetics. 1974;76:537-549.
15.Takahashi S, Fukamizu A, Hatae T, Yamada
Y, Sugiyama F, Kajiwara N, Yagami K, Murakami K. Species-specific
kinetics of mouse renin contribute to maintenance of normal blood
pressure in transgenic mice with overexpressed human
angiotensinogen. J Vet Med Sci. 1992;54:1191-1193.
16.Fukamizu A, Sugimura K, Takimoto E,
Sugiyama F, Seo MS, Takahashi S, Hatae T, Kajiwara N, Yagami K,
Murakami K. Chimeric renin-angiotensin system demonstrates
sustained increase in blood pressure of transgenic mice carrying
both human renin and human angiotensinogen genes. J Biol Chem.
1993;268:11617-11621.
17. Huang PL, Huang Z, Mashimo H, Bloch KD,
Moskowitz MA, Bevan JA, Fishman MC. Hypertension in mice lacking
the gene for endothelial nitric oxide synthase [see comments]. Nature.
1995;377:239-242.
18.Kuro-o M, Hanaoka K, Hiroi Y, Noguchi T,
Fujimori Y, Takewaki S, Hayasaka M, Katoh H, Miyagishi A, Nagai
R, et al. Salt-sensitive hypertension in transgenic mice
overexpressing Na(+)- proton exchanger. Circ Res.
1995;76:148-153.
19.Lopez MJ, Wong SK, Kishimoto I, Dubois
S, Mach V, Friesen J, Garbers DL, Beuve A. Salt-resistant
hypertension in mice lacking the guanylyl cyclase-A receptor for
atrial natriuretic peptide. Nature. 1995;378:65-68.
20. Ohkubo H, Kawakami H, Kakehi Y, Takumi
T, Arai H, Yokota Y, Iwai M, Tanabe Y, Masu M, Hata J, et al.
Generation of transgenic mice with elevated blood pressure by
introduction of the rat renin and angiotensinogen genes. Proc
Natl Acad Sci U S A. 1990;87:5153-5157.
21. Veress AT, Chong CK, Field LJ,
Sonnenberg H. Blood pressure and fluid-electrolyte balance in
ANF-transgenic mice on high- and low-salt diets. Am J Physiol.
1995;269:R186-192.
22.Wang J, Xiong W, Yang Z, Davis T, Dewey
MJ, Chao J, Chao L. Human tissue kallikrein induces hypotension
in transgenic mice. Hypertension. 1994;23:236-243.
23. Qiu Y, Rizvi A, Tang XL, Manchikalapudi
S, Takano H, Jadoon AK, Wu WJ, Bolli R. Nitric oxide triggers
late preconditioning against myocardial infarction in conscious
rabbits. Am J Physiol. 1997;273:H2931-2936.
24. Takano H, Manchikalapudi S, Tang XL,
Qiu Y, Rizvi A, Jadoon AK, Zhang Q, Bolli R. Nitric oxide
synthase is the mediator of late preconditioning against
myocardial infarction in conscious rabbits. Circulation.
1998;98:441-449.
25. Marber MS, Latchman DS, Walker JM,
Yellon DM. Cardiac stress protein elevation 24 hours after brief
ischemia or heat stress is associated with resistance to
myocardial infarction. Circulation. 1993;88:1264-1272.
26. Baxter GF, Marber MS, Patel VC, Yellon
DM. Adenosine receptor involvement in a delayed phase of
myocardial protection 24 hours after ischemic preconditioning. Circulation.
1994;90:2993-3000.
27. Baxter GF, Goma FM, Yellon DM.
Characterisation of the infarct-limiting effect of delayed
preconditioning: timecourse and dose-dependency studies in rabbit
myocardium. Basic Res Cardiol. 1997;92:159-167.
28. Baxter GF, Goma FM, Yellon DM.
Involvement of protein kinase C in the delayed cytoprotection
following sublethal ischemia in rabbit myocardium. Br J
Pharmacol. 1995;115:222-224.